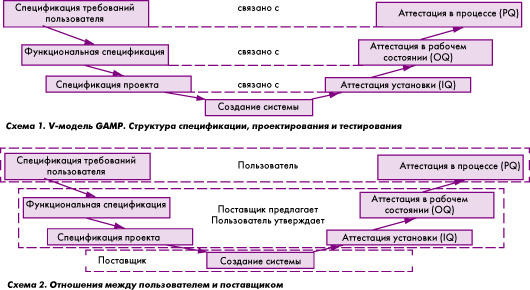








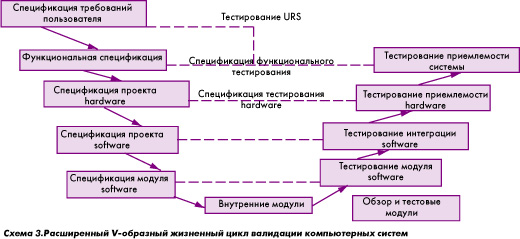
Категория: Бланки/Образцы
1. Введение
2. Предмет и подход к валидации
3. Валидационная комиссия
4. Документация
5. Список машин и оборудования подлежащих квалификации.
6. Валидация чистых помещений.
7. Валидация котлов.
8. Валидация перерабатывающего оборудования.
9. Валидация машины для розлива / наполнения туб.
10. Валидация системы подготовки чистой воды.
Чтобы скачать файл: "Валидационный мастер-план",
необходима регистрация с последующей авторизацией.
Автор: Римма Еременко, ФК "Здоровье"
Версия: pdf Размер файла: 238.92 Kb
Мы разрешаем использовать, цитировать, копировать, транслировать и переводить любые наши материалы в сети Интернет
при условии установки прямой ссылки на этот конкретный материал на сайте Klub OK .net
Для того чтобы опубликовать свой материал (статью, книгу и т.д.),
вам достаточно направить его по адресу klubok@klubok.net
в любом удобном вам формате.
Copyright © 2003-2016 Klub OK .net. Андрей Гарин








Автор: Александр В. Александров. тезисы доклада для 13-го международного проекта "Созвездие качества'2012"
Сложности интерпретации требований ИСО 9001 в отношении «валидации специальных процессов» известны каждому менеджеру по качеству, который хоть раз сталкивался с внедрением этого стандарта. Вот и получается на практике, что вполне стандартное требование, четко и подробно описанное в нормативных документах для фармацевтической отрасли, для других отраслей сопровождается пугающим количеством толкований и разъяснений. Выдержка из ИСО 9001, пункт 7.5.2: «Организации следует осуществлять валидацию любого процесса выпуска продукции или предоставления услуг, подтверждение (верификацию) конечного выхода которого невозможно провести последующим мониторингом или измерениями, и следовательно, недостатки которого (т.е. конечного выхода) выявляются только после начала использования продукта или завершения оказания услуги». И соответственно в стандарте ИСО 9000:2005 в 3-м примечании к определению термина «процесс» (3.4.1) указано, что «Процесс, в котором подтверждение (верификация) соответствия конечной продукции не может быть проведено своевременно или влечет к значительным экономическим затратам, часто называют специальным».
Для фармацевтической отрасли под определение специального процесса полностью подпадает «технологический процесс», т.е. процесс изготовления лекарственного препарата. Естественно предполагается, что препарат качественный. Что такое «качество лекарственного препарата»? Прежде всего, это его эффективность, безопасность и соответствие спецификации (стандарту качества). Соответствие спецификации можно подтвердить контролем качества (по сути, верификацией), однако проблема в том, что контроль выборочный. Т.е. результаты контроля распространяются на всю серию на основании тестирования образцов, которые не идут в продажу. И это еще большая задача, доказать что выборка – репрезентативна. Дальше еще хуже. Безопасность и эффективность лекарственного препарата подтверждаются (или не подтверждаются) только в процессе его применения– т.е. тогда, когда что-то изменить, исправить уже невозможно.
Именно поэтому одним из ключевых принципов GMP (GoodManufacturingPractice, Надлежащая производственная практика) считается валидация технологического процесса. Валидации процессов посвящено отдельное приложение – Приложение 15 GMP, которое было включено в GMP в 1987 году. И что очень важно, без результатов валидации коммерческий выпуск лекарственного препарата невозможен, правильнее сказать жестче – запрещен. Валидация поддерживает концепцию GMP в переносе центра тяжести с контроля качества готового продукта на обеспечение качества процесса. Кроме того, процедуры организации и проведения валидации отражают базовые принципы GMP, а именно: продуманное планирование, четкое выполнение и подробное документирование. Валидация включает такие важные для GMP элементы, как научный подход на основе оценки рисков по качеству и управление изменениями.
Корень «валид» означает пригодный. В русском языке есть несколько слов с этим корнем, например «инвалид» - непригодный, «валидный» - пригодный. В фармацевтической отрасли, термин «валидация» трактуется следующим образом: «Процесс документированного подтверждения достижения разумной степени уверенности в том, что
По сути, валидация технологического процесса – это конечная цель, для достижения которой нужно последовательно провести валидацию ряда других связанных процессов. В GMP общий термин «валидация» разделяется на два понятия: «валидация процессов» и «квалификация производственных систем». Квалификация производственных систем – это часть валидации процесса, направленная на документальное подтверждение пригодности оборудования, инженерных систем, комплекса помещений, которые используются в производстве лекарственного препарата. Квалификация проводится для того, чтобы быть уверенными в том, что производственная система не влияет на качество продукта, а также для того, что если мы в ходе непосредственно валидации технологического процесса получим негативный результат – это не может быть связано с отказами оборудования/систем, а причины нужно искать в самом технологическом процессе). По своей логике, квалификация производственных систем – это некая предупреждающая мера.

Таким образом, под валидацией процесса в фармацевтической отрасли подразумевается*.
Организация валидационных работ
Ответственность за проведение валидационныхработ как правило возложена на Отдел обеспечения качества. Для координации деятельности структурных подразделений создается Валидационная комиссия и валидационные группы.
Стандартный пакет валидационной документации:
Для каждого критического объекта инфраструктуры должна быть проведена квалификация, которая, как правило, осуществляется в четыре последовательных этапа:
Квалификация проекта (DQ) направлена на документированное подтверждение пригодности проекта (конструкции, проектного решения) технических средств, инженерных систем и оборудования для их предполагаемого использования. Объем работ на этом этапе:
Квалификация монтажа (IQ) направлена на документированное подтверждение того, что технические средства, инженерные системы и оборудование сконструированы, оснащены и смонтированы в соответствии с рабочей документацией проекта и рекомендациями производителя. Объем работ на этом этапе:
Квалификация функционирования (OQ) направлена на документированное подтверждение того, что технические средства, инженерные системы и оборудование функционируют должным образом по всему заявленному диапазону рабочих характеристик. Объем работ на этом этапе:
Как правило, после этого этапа квалификации объект вводится в эксплуатацию.
Квалификация функционирования (PQ) проводится для инженерных систем, которые работают непрерывно, а также для оборудования со сложным управлением.Квалификация в эксплуатации – это документированное подтверждение того, что технические средства, инженерные системы и оборудование при совместном (или длительном) использовании могут надежно функционировать с получением воспроизводимых свойств продукта.
При этом, если производственная система оснащена автоматизированной системой мониторинга параметров, или обработки данных, дополнительно должна проводиться валидация компьютеризированной системы.
Валидация аналитических методик
Каждая аналитическая и микробиологическая методика, которая используется для контроля качества сырья, полупродукта или готового продукта должна пройти валидацию. Это означает, что мы обязаны получить доказательства пригодности такой методики для контроля конкретного продукта и соответственно, гарантии получения достоверных результатов. В этом плане, требования GMP полностью совпадают с требованиями ИСО 17025.
Процедуры очистки оборудования должны также пройти валидацию до того, как мы приступим к производству препарата на этом оборудовании. Прежде всего, эта валидация направлена на получение гарантий возможности проведения качественной очистки после изготовления такого продукта. По сути, это минимизация риска перекрестного загрязнения при переходе на производство другого продукта на этом же оборудовании. Если на оборудовании останутся остатки предыдущего продукта, это не будет обнаружено – так как отсутствует аналитический контроль именно на наличие таких примесей.
Валидация асептических условий
При производстве стерильных лекарственных средств с использованием асептических технологий до начала самого технологического процесса необходимо подтвердить, что на всем протяжении процесса изготовления препарата (т.е. длительность процесса), в продукт не попадает ни один микроорганизм. Валидация асептических условий проводится по сценарию имитации с помощью питательных сред.
Валидация технологического процесса
И непосредственно, валидация каждого из этапов технологического процесса проводится на 3-х последовательных сериях с учетом «наихудшего случая». И, что очень важно, валидациятехнологического процесса проводится отдельно для каждого продукта и его заявляемого размера серии. Наихудший случай – это проведение процесса при таких условиях и обстоятельствах (для параметров процесса, режимов работы оборудования), которые имеют максимальные шансы вызвать отклонение процесса или несоответствие продукта по сравнению с идеальными условиями. Логика очень проста – если при таких условиях мы получаем качественный продукт, значит, гарантированно мы будем достигать качества внутри заданных диапазонов.
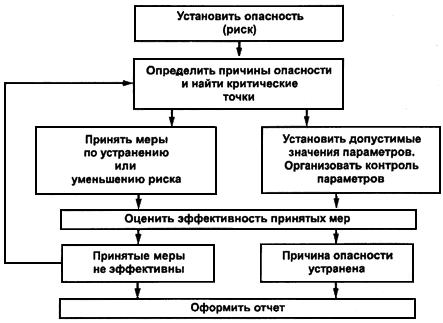
Через заданные периоды эксплуатации (использования), каждый объект/процесс должны пройти повторнуювалидацию. Основная цель повторной валидации (ревалидации) – это получить подтверждения того, что объект/процесс продолжает находиться в валидном состоянии.Это полностью отражает логику GMP: «Для подтверждения качества продукта недостаточно провести валидацию в начале его жизненного цикла, необходимо обеспечить мониторинг и постоянное улучшение» (см. схему ниже).
Рассматривается плановая и внеплановая ревалидация. Плановая – проводится по графикув соответствии с заранее установленной периодичностью (как правило, через 12-24 мес.). Внеплановаяревалидация – после длительных простоев, при появлении тренда отклонений или при внесении изменений.

* Перечень приведен в той последовательности, в которой должны проводиться валидационные работы
18-20 октября 2016 года в ВЦ «КиевЭкспоПлаза» состоится VII Международная выставка оборудования и технологий для фармацевтической промышленности PHARMATechExpo – единственная в Украине выставка, в рамках которой представлен весь процесс фармацевтического производства: от разработки субстанций и контроля качества сырья, оборудования для производства фармацевтических препаратов и упаковочных технологий до транспортировки, хранения лекарственных средств и подбора персонала.

Цель любого предприятия – поставка на рынок продукции, процессы производства которой являются устойчивыми и воспроизводимыми, исключающими выпуск несоответствующей продукции и материальные потери.
Важный инструмент для достижения этой цели – валидация процессов и оборудования, которая является одним из основных элементов системы обеспечения качества.
Валидационный мастер-план обобщает концепцию предприятия в отношении валидации, определяет ответственность персонала, устанавливает перечень объектов, подлежащих валидации, периодичность проведения и требования к документальному оформлению валидации.
Объекты и объем валидации, определенные в валидационном мастер-плане. устанавливаются на основании всестороннего анализа рисков, целью которого является защита потребителя от использования некачественной продукции.
Валидационный мастер-план составляется на основании нормативных требований и с учетом специфических особенностей предприятия.
13-14 декабря 2016
Семинар: «Валидация компьютеризированных систем в фармацевтической деятельности. Как обеспечить регуляторное соответствие требованиям GMP на практике?»
19 октября 2016
Семинар: «Аттестация складских помещений, холодильных камер, холодовых цепочек. Практические аспекты, примеры»
21 октября 2015
Семинар «Валидация компьютеризированных систем. Как обеспечить регуляторное соответствие на практике?»
Сложности интерпретации требований ИСО 9001 в отношении «валидации специальных процессов» известны каждому менеджеру по качеству, который хоть раз сталкивался с внедрением этого стандарта. Вот и получается на практике, что вполне стандартное требование, четко и подробно описанное в нормативных документах для фармацевтической отрасли, для других отраслей сопровождается пугающим количеством толкований и разъяснений. Выдержка из ИСО 9001, пункт 7.5.2: «Организации следует осуществлять валидацию любого процесса выпуска продукции или предоставления услуг, подтверждение (верификацию) конечного выхода которого невозможно провести последующим мониторингом или измерениями, и следовательно, недостатки которого (т.е. конечного выхода) выявляются только после начала использования продукта или завершения оказания услуги». И соответственно в стандарте ИСО 9000:2005 в 3-м примечании к определению термина «процесс» (3.4.1) указано, что «Процесс, в котором подтверждение (верификация) соответствия конечной продукции не может быть проведено своевременно или влечет к значительным экономическим затратам, часто называют специальным» .

Александр В. Александров, президент Группы компаний ВИАЛЕК
Для фармацевтической отрасли под определение специального процесса полностью подпадает «технологический процесс», т.е. процесс изготовления лекарственного препарата. Естественно предполагается, что препарат качественный. Что такое «качество лекарственного препарата»? Прежде всего, это его эффективность. безопасность и соответствие спецификации (стандарту качества). Соответствие спецификации можно подтвердить контролем качества (по сути, верификацией), однако проблема в том, что контроль выборочный. Т.е. результаты контроля распространяются на всю серию на основании тестирования образцов, которые не идут в продажу. И это еще большая задача, доказать что выборка – репрезентативна. Дальше еще хуже. Безопасность и эффективность лекарственного препарата подтверждаются (или не подтверждаются) только в процессе его применения – т.е. тогда, когда что-то изменить, исправить уже невозможно.
Именно поэтому одним из ключевых принципов GMP ( G ood M anufacturing P ractice, Надлежащая производственная практика ) считается валидация технологического процесса. Валидации процессов посвящено отдельное приложение – Приложение 15 GMP. которое было включено в GMP в 1987 году. И что очень важно, без результатов валидации коммерческий выпуск лекарственного препарата невозможен, правильнее сказать жестче – запрещен. Валидация поддерживает концепцию GMP в переносе центра тяжести с контроля качества готового продукта на обеспечение качества процесса. Кроме того, процедуры организации и проведения валидации отражают базовые принципы GMP, а именно: продуманное планирование, четкое выполнение и подробное документирование. Валидация включает такие важные для GMP элементы, как научный подход на основе оценки рисков по качеству и управление изменениями.
Что такое валидация?
Корень «валид» означает пригодный. В русском языке есть несколько слов с этим корнем, например «инвалид» — непригодный, «валидный» — пригодный. В фармацевтической отрасли, термин «валидация» трактуется следующим образом: «Процесс документированного подтверждения достижения разумной степени уверенности в том, что
— Производственный процесс,
— Аналитические методики,
— Используемое оборудование,
— Производственные системы,
соответствуют действующим принципам GMP и выполняют свое функциональное назначение, т.е. их использование действительно дает ожидаемые результаты ».
По сути, валидация технологического процесса – это конечная цель, для достижения которой нужно последовательно провести валидацию ряда других связанных процессов. В GMP общий термин «валидация» разделяется на два понятия: «валидация процессов» и «квалификация производственных систем». Квалификация производственных систем – это часть валидации процесса, направленная на документальное подтверждение пригодности оборудования, инженерных систем, комплекса помещений, которые используются в производстве лекарственного препарата. Квалификация проводится для того, чтобы быть уверенными в том, что производственная система не влияет на качество продукта, а также для того, что если мы в ходе непосредственно валидации технологического процесса получим негативный результат – это не может быть связано с отказами оборудования/систем, а причины нужно искать в самом технологическом процессе). По своей логике, квалификация производственных систем – это некая предупреждающая мера.
Схема – Три этапа валидации технологического процесса
Таким образом, под валидацией процесса в фармацевтической отрасли подразумевается [1] :
— Квалификация чистых помещений
— Квалификация инженерных систем (подготовка чистого воздуха, воды очищенной и воды для инъекций, сжатого воздуха и т.п.)
— Квалификация производственного оборудования
— Квалификация аналитического оборудования (используемого для контроля качества сырья, полупродуктов и готовой продукции)
— Квалификация складских зон (сырье, готовая продукция)
— Валидация компьютеризированных систем, включая квалификацию ИТ-инфраструктуры
— Валидация аналитических методик
— Верификация фармакопейных методик (методики, внесенные в национальную или региональные Фармакопеи )
— Квалификация в эксплуатации (PQ. P erformance Qualification)
Квалификация проекта (DQ) направлена на документированное подтверждение пригодности проекта (конструкции, проектного решения) технических средств, инженерных систем и оборудования для их предполагаемого использования. Объем работ на этом этапе:
— Описание системы (функция, параметры оборудования, особые характеристики)
— Техническая документация (нормативные требования, документация по оборудованию)
— Оценка конструкции (конструкционные материалы, оценка риска загрязнений)
— Компоненты/элементы оборудования/системы
— Анализ возможных отказов/дефектов
— Анализ способа изготовления (критические параметры работ при изготовлении оборудования, требования по калибровке)
Квалификация монтажа (IQ) направлена на документированное подтверждение того,
что технические средства, инженерные системы и оборудование сконструированы, оснащены и смонтированы в соответствии с рабочей документацией проекта и рекомендациями производителя. Объем работ на этом этапе:
— Наличие достаточной документации
— Наличие всех элементов в поставке
— Правильность монтажа и подключений
— Соответствие контактирующих материалов
— Соответствие средств измерений
Квалификация функционирования (OQ) направлена на документированное подтверждение того, что технические средства, инженерные системы и оборудование функционируют должным образом по всему заявленному диапазону рабочих характеристик. Объем работ на этом этапе:
— Приемлемость документации (инструкции по эксплуатации, обслуживанию);
— Испытания, включающие условие или ряд условий, охватывающих верхний и нижний пределы рабочих параметров:
— Срабатывание блокировок/сигнализаций.
Как правило, после этого этапа квалификации объект вводится в эксплуатацию.
Квалификация функционирования (PQ ) проводится для инженерных систем, которые работают непрерывно, а также для оборудования со сложным управлением. Квалификация в эксплуатации – это документированное подтверждение того, что технические средства, инженерные системы и оборудование при совместном (или длительном) использовании могут надежно функционировать с получением воспроизводимых свойств продукта .
При этом, если производственная система оснащена автоматизированной системой мониторинга параметров, или обработки данных, дополнительно должна проводиться валидация компьютеризированной системы .
Валидация аналитических методик
Каждая аналитическая и микробиологическая методика, которая используется для контроля качества сырья, полупродукта или готового продукта должна пройти валидацию. Это означает, что мы обязаны получить доказательства пригодности такой методики для контроля конкретного продукта и соответственно, гарантии получения достоверных результатов. В этом плане, требования GMP полностью совпадают с требованиями ИСО 17025.
Процедуры очистки оборудования должны также пройти валидацию до того, как мы приступим к производству препарата на этом оборудовании. Прежде всего, эта валидация направлена на получение гарантий возможности проведения качественной очистки после изготовления такого продукта. По сути, это минимизация риска перекрестного загрязнения при переходе на производство другого продукта на этом же оборудовании. Если на оборудовании останутся остатки предыдущего продукта, это не будет обнаружено – так как отсутствует аналитический контроль именно на наличие таких примесей.
Валидация асептических условий
При производстве стерильных лекарственных средств с использованием асептических технологий до начала самого технологического процесса необходимо подтвердить, что на всем протяжении процесса изготовления препарата (т.е. длительность процесса), в продукт не попадает ни один микроорганизм. Валидация асептических условий проводится по сценарию имитации с помощью питательных сред.
Валидация технологического процесса
И непосредственно, валидация каждого из этапов технологического процесса проводится на 3-х последовательных сериях с учетом «наихудшего случая». И, что очень важно, валидация технологического процесса проводится отдельно для каждого продукта и его заявляемого размера серии. Наихудший случай – это проведение процесса при таких условиях и обстоятельствах (для параметров процесса, режимов работы оборудования), которые имеют максимальные шансы вызвать отклонение процесса или несоответствие продукта по сравнению с идеальными условиями. Логика очень проста – если при таких условиях мы получаем качественный продукт, значит, гарантированно мы будем достигать качества внутри заданных диапазонов.
Через заданные периоды эксплуатации (использования), каждый объект/процесс должны пройти повторную валидацию. Основная цель повторной валидации (ревалидации) – это получить подтверждения того, что объект/процесс продолжает находиться в валидном состоянии. Это полностью отражает логику GMP: «Для подтверждения качества продукта недостаточно провести валидацию в начале его жизненного цикла, необходимо обеспечить мониторинг и постоянное улучшение» (см. схему ниже).
Схема – Надзор над валидированным объектом/процессом
Рассматривается плановая и внеплановая ревалидация. Плановая – проводится по графику в соответствии с заранее установленной периодичностью (как правило, через 12-24 мес.). Внеплановая ревалидация – после длительных простоев, при появлении тренда отклонений или при внесении изменений.
[1] Перечень приведен в той последовательности, в которой должны проводиться валидационные работы
13-й международный проект «Созвездие качества’2012»
Александр В. Александров, Группа компаний ВИАЛЕК








